The way in which DNA methylation on a genomic scale, aka the Methylome, gets established, maintained and sometimes altered has profound implications for biological research and understanding, and may someday lead to advanced therapies for a variety of diseases. Olivia May’s article delivers more details about the inner working of the Methylome, and what it may mean to future research:
DNA Methylation: Establishing the Methylome
Throughout our lifespan, chemical changes subtly occur in our DNA. A comparison of the DNA of a newborn baby with that of a centenarian shows that the scope of these changes can be dramatic, potentially having an influence on disease.1
This may help explain why our risk of cancer and other diseases increases as we get older. Such exogenous influences can also be inherited, impacting the genetics of an individual’s offspring. Epigenetic regulation involves genetic control by factors other than an individual’s DNA sequence.
These heritable changes alter DNA accessibility and chromatin structure, thereby regulating patterns of gene expression. Through epigenetic programming, genes are switched on or off as needed to determine which proteins are transcribed and expressed.
This process is crucial to normal development (controlling genomic imprinting and X-chromosome inactivation) as well as for the suppression of transposable elements and the maintenance of stable cellular identities. Unfortunately, changes in DNA methylation patterns also contribute to human diseases, like cancer, for which risk increases with age.
Download this full article, DNA Methylation: Establishing & Editing the Methylome by Olivia L. May, Ph.D compliments of our friends at Cayman Chemical.
DNA methylation, the chemical process that adds a methyl group to DNA, is the principal manner in which genes are transcriptionally repressed or silenced. This modification occurs specifically at CpG sites, regions in which a cytosine nucleotide is located next to a guanine nucleotide that is linked by a phosphate (Figure 1).
Inserting methyl groups changes the appearance and structure of DNA, which may directly block DNA recognition and binding of transcription factors, or may attract other factors that preferentially bind to DNA to interfere with transcription factor accessibility.
Three families of proteins which bind methylated DNA have been identified so far.2,3 These include MBD domain proteins, Kaiso and Kaiso-like proteins, and SRA domain proteins. By recruiting these proteins, DNA methylation marks can promote the persistence of certain histone states, such as deacetylation, thus enabling posttranslational histone modifications.
As an example, methyl CpG-binding domain protein 2 (MeCP2), a member of the MBD family, binds to methyl CpG and recruits HDACs, which promote chromosome condensation and transcriptional repression.
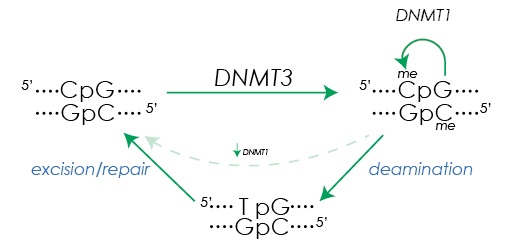 Figure 1. DNA methylation and demethylation. De novo DNA methylation at the cytosine in CpG dinucleotides is initiated by DNMT3A and DNMT3B. After replication, DNMT1 maintains the methylation state in the daughter strands. Base excision repair mechanisms facilitate removal of methylation after deamination of methyl cytosine (5mC) creates a T:G mismatch.
Figure 1. DNA methylation and demethylation. De novo DNA methylation at the cytosine in CpG dinucleotides is initiated by DNMT3A and DNMT3B. After replication, DNMT1 maintains the methylation state in the daughter strands. Base excision repair mechanisms facilitate removal of methylation after deamination of methyl cytosine (5mC) creates a T:G mismatch.
Establishing Methylation Marks
CpG sites are methylated by one of three DNA methyltransferases (DNMTs). During embryogenesis, de novo methylation is typically performed by DNMT3A and DNMT3B. Both have similar structures, comprising a PWWP domain, PHD-like or ADD domain, and a carboxy-terminal catalytic domain. The PWWP domains are necessary for binding of DNMT3A and DNMT3B to chromatin in vivo. DNMT3L, which lacks the PWWP and a functional catalytic domain, forms a heterotetramer with DNMT3A or DNMT3B and stimulates the activity of its de novo partners, guiding the recognition of DNA targets.4 The PHD domain of DNMT3L interacts with the animo terminal tail of histone H3 and is activated only when bound to unmethylated DNA.5
Appropriate combinations of histone modifications create either protective or permissive conditions for the docking of de novo methylation complexes. Specific methylation of the lysine at residue 4 (H3K4) by SETD1 inhibits DNMT3L binding. H3K4 methylation is a marker for active genes, and there is an inverse correlation between the presence of trimethylated H3K4 and DNA methylation at promoters.5,6
Indeed, chromatin at the majority of unmethylated CpGS is enriched in H3K4 di- and tri-methylation. Removal of H3K4 methylation by the histone demethylase LSD2 (KDM1B) is required for establishment of DNA methylation at the promoters of certain imprinted genes.
Additionally, DNMT3A binding is promoted by SETD2 trimethylation of lysine 36 of histone 3 (H3K36me3), whereas the H3K36me2 demethylase KDM2A binds to unmethylated CpGs resulting in depletion of H3K36me2.6 Many questions still remain however as to where and how patterns of methylation are established to target appropriate, region-specific histone modifications.7
You can find the entire article, DNA Methylation: Establishing & Editing the Methylome by Olivia L. May, Ph.D. on the Cayman Chemical Website.
References
- Heyn, H., Li, N., Ferreira, H.J., et al. Proc. Natl. Acad. Sci. 109, 10522-10527 (2012).
- Klose, R.J. and Bird, A.P. Trends Biochem. Sci. 31(2), 89-97 (2006).
- Clouaire, T., de las Heras, J.I., Merusi, C. et al. Nucleic Acids Res. 38(14), 4620-4634 (2010).
- Chédin, F., Lieber, M.R., and Hsieh, C.L. Proc. Natl. Acad. Sci. 99(26), 16916-16921 (2002).
- Kelsey, G. Cell Res. 21, 1155-1156 (2011).
- Kelsey, G. and Feil, R. Phil. Trans. R. Soc. B 368, 20110336 (2012).
- Bird, A. Genes Dev. 16, 6-21 (2002).