Location is important. But while we have great GPS tools to locate ourselves in the macro-world, microscopic things can be a bit harder to find. You might think it would be tough to lose something in as tiny a space as the inside of a cell, but that’s what I thought about my keys in my pocket. Fortunately, two recent techniques show how to better keep track of RNA and proteins inside live cells.
Split Fluorescent Proteins For Better Protein Labeling
Fluorescent proteins are pretty great (Nobel-worthy, even) ways to find a protein inside a cell. However, they can be fairly bulky, so some people are moving toward small epitope tags. These are not fluorescent on their own, but they bind and localize some other fluorescent molecule. This is great for minimally intrusive genetic labeling, but it tends to suffer from high background fluorescence from unbound molecules. A nice solution to the background problem is split GFP, which was developed in Geoffrey Waldo’s lab in 2005. A tiny sliver of super-folder GFP (sfGFP11) can be split off from the main barrel of the protein (sfGFP1-10), and although the two halves are not fluorescent, if they find each other, they’ll reassemble and recover fluorescence.
The tiny size of sfGFP11 makes it a very nice epitope tag, as several studies have shown. Now, Daichi Kamiyama and Sayaka Sekine, working with Bo Huang at UC San Francisco, have expanded this as a general method for multiple epitope tagging applications.
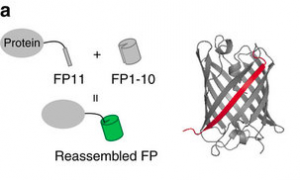
The FP11 sliver is a svelte and nimble genetic epitope, but it sticks into the main FP1-10 barrel to localize full GFP, YFP, or CFP fluorescence. Image via Kamiyama et al, Nature Communications 2016.
In particular, Daichi and Sayaka found that:
- Reassembled split sfGFP was as bright as the full-length version with very low background, and the small sfGFP11 epitope worked well for creating libraries of fluorescently labeled genes.
- The Y66W and T203Y mutations to make sfGFP into CFP and YFP also worked well, enabling multi-color imaging.
- Split sfCherry worked too, but it was not as bright as full-length sfCherry.
- Tandem repeats of the FP11 sliver made labeling proportionally brighter, allowing better visualization at lower light intensity.
In a final clever trick for better CRISPR translational activation, the team fused tandem repeats of sfGFP11 to dCas9 and then fused sfGFP1-10 to the VP64 activation domain. When they targeted this to a promoter region with an appropriate sgRNA, the target gene expression increased 45x – much more than it would with just a single VP64 domain.
CRISPR/Cas9 for RNA Tracking in Live Cells
Fluorescent protein fusions are great for tagging proteins, but RNAs are still pretty hard to follow inside cells. Fortunately, David Nelles, working in the lab of Gene Yeo at UC San Diego, has a clever way to find RNAs with CRISPR/Cas9.
Cas9 normally binds DNA, but it can be tricked into binding RNA if you give it a synthetic, PAM-containing oligo (called a PAMmer), made of a DNA PAM and stabilized RNA bases that hybridizes with the target RNA. This makes the target look like a double-stranded helix with a DNA PAM. This has been done in vitro, but this team showed it works in vivo with native mRNAs.
The team used FISH to verify that RNA-targeting dCas9 (RCas9) really did co-localize with the target mRNA. By adding a GFP tag to RCas9, they could follow mRNAs inside live cells in real time.
To firm up your own sense of place in the world, check out these two fluorescent localization papers in Nature Communications, 2016, and Cell, 2016.