You’ve read the NOMe-seq (Nucleosome Occupancy and Methylome sequencing) papers. Seen the splashy advertisements. Now, we’ve consulted with the experts at Active Motif to get the inside scoop on how to get started, and get the most out of your NOMe-seq experience.
NOMe-Seq was originally published and developed by researchers in the Peter Jones lab at USC, and is the first technique to simultaneously analyze nucleosome occupancy and CpG methylation status on the same DNA strand. NOMe-seq enables investigation of relationships between nucleosome occupancy, transcription factor binding and DNA methylation within the same DNA molecule, even providing a temporal snap shot of the relationship between nucleosome occupancy and DNA methylation. The information that NOMe-seq generates can be used to:
- Find very subtle changes in nucleosome position
- Determine the presence of non-nucleosomal DNA binding proteins (transcription factors)
- Identify novel imprinted genes
- Study the effect of various chromatin states on gene regulation
- Determine whether the presence of a particular SNP correlates with chromatin structure
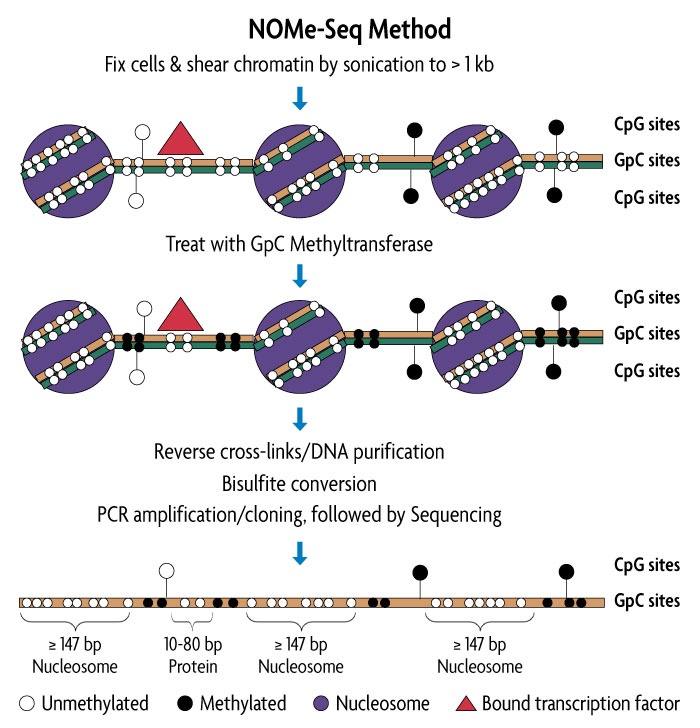
The NOMe-Seq protocol preserves protein/DNA interactions and maintains endogenous CpG methylation states and can work on less than 1 million cells. While traditional methods are only able to scope out one epigenetic modification at a time, these attributes give NOMe-seq the huge advantage of assessing both DNA methylation and high-resolution nucleosome position, and their relationship to each other, in one experiment.
NOMe-Seq Tips and Tricks
We caught up with Dr. Terry Kelly, Active Motif R&D Manager, and co-inventor of the NOMe-Seq method, to pick her brain about the best ways to get started, and generate the best results out of a NOMe-Seq run. Here is her advice:
Double Check Nuclei Lysis
Complete lysis of the nucleus will ensure that the methyltransferase has access to the chromatin.
Make Use of Test Regions
Kelly suggests that you should “Verify successful enzyme treatment on test regions that are expected to be nucleosome depleted (accessible) and nucleosome occupied (inaccessible). For example active and silent gene promoters.”
Include “No Methyltransferase” Controls
“GCG motifs should be excluded from analyses, since you won’t know if they are methylC because of the CG and the GC context, but including a no methyltransferase control sample can help you figure out if the CG site is endogenously methylated.” Says Kelly.
Start Off Small
Kick off a project by sequencing 5-10 molecules of a particular region. Kelly advises that, “If the molecules have the same pattern you may not need to sequence more, since having 50 molecules will likely give you the same information as 5.”
Size Matters
The longer amplicons are, the better. For locus specific PCR, amplicons over 700 base pairs can be difficult to amplify, but ones that are at least 350bp should enable you to clearly see multiple nucleosomes or distinguish between linker regions and nucleosome depleted regions (NDRs). Kelly has also noticed that “When going genome-wide the longer reads are definitely better, especially if you can do paired end analysis. With paired ends you, hopefully, can have reads that overlap, essentially giving you more than 1 nuclesosme’s length and helping distinguish between what is a transcription factor (TF) and what is a nucleosome”.
Content is King
For optimum results, PCR primers need to be designed to avoid CG or GC dinucleotides. Those motifs could be altered during the bisulfite conversion process, causing variable PCR amplification efficiency. That could bias your data and wreak havoc for analysis. Active Motif also suggests that you might want to include spike-ins with known ratios of methylated and unmethylated amplicons to check for PCR bias.
You can find more information on NOMe-Seq Kits at the Active Motif website.