During this time of the year, it’s easy to get caught up in landscaping. Regardless of whether your landscaping is of the epigenetic or garden variety, it seems that either way we can all benefit from the wisdom of the old saying, “can’t see the forest for the trees”. Sometimes we just get so caught up in the details that we forget to take a step back and consider the bigger picture.
Thankfully, the lab of Shiva Singh at the University of Western Ontario bring forth their ‘forest city’ perspective and offer-up an integrated analysis of fetal alcohol exposure. Instead of examining the relationship of one epigenetic methylation to gene expression, they decided to throw DNA methylation, two histone methylations, and ncRNA into the mix. Also, if that wasn’t enough to impress you, then maybe it’s worth mentioning that it’s the first report of genome-wide alterations to histone methylation resulting from fetal alcohol exposure.












The Epigenomics of Fetal Alcohol Exposure
Previously, the Singh lab has shown that moderate prenatal alcohol exposure produces long-term alterations to DNA methylation and ncRNA in the whole brains of exposed mice grown to adulthood. They’ve also shown that some of these alterations to DNA methylation are present in the saliva of young human children with FASD. Now, they’ve taken on histone methylation and the brain’s hippocampus, which is a specific region central to learning and memory.
In their current model, the team injected mice with alcohol on postnatal days 4 and 7, which is neurodevelopmentally equivalent to the third trimester of human pregnancy. They then raised the mice into adulthood (postnatal day 70) and utilized gene expression arrays as well as promoter arrays for DNA methylation, H3K4me3, and H3K27me3.
Here’s what they found:
- Long lasting gene expression, DNA methylation, and histone methylation changes across the whole genome.
- An enrichment of similar but distant genes, associative networks, and canonical pathways related to oxidative stress across all analyses.
- The differential gene expression benefited from the use of droplet digital PCR (ddPCR) technology, which enabled the confirmation of the low-fold change and/or low abundance transcripts that were beyond the detection limits of standard qPCR.
- Finally, the differential methylation was also confirmed by the pyrosequencing experts at EpigenDx.
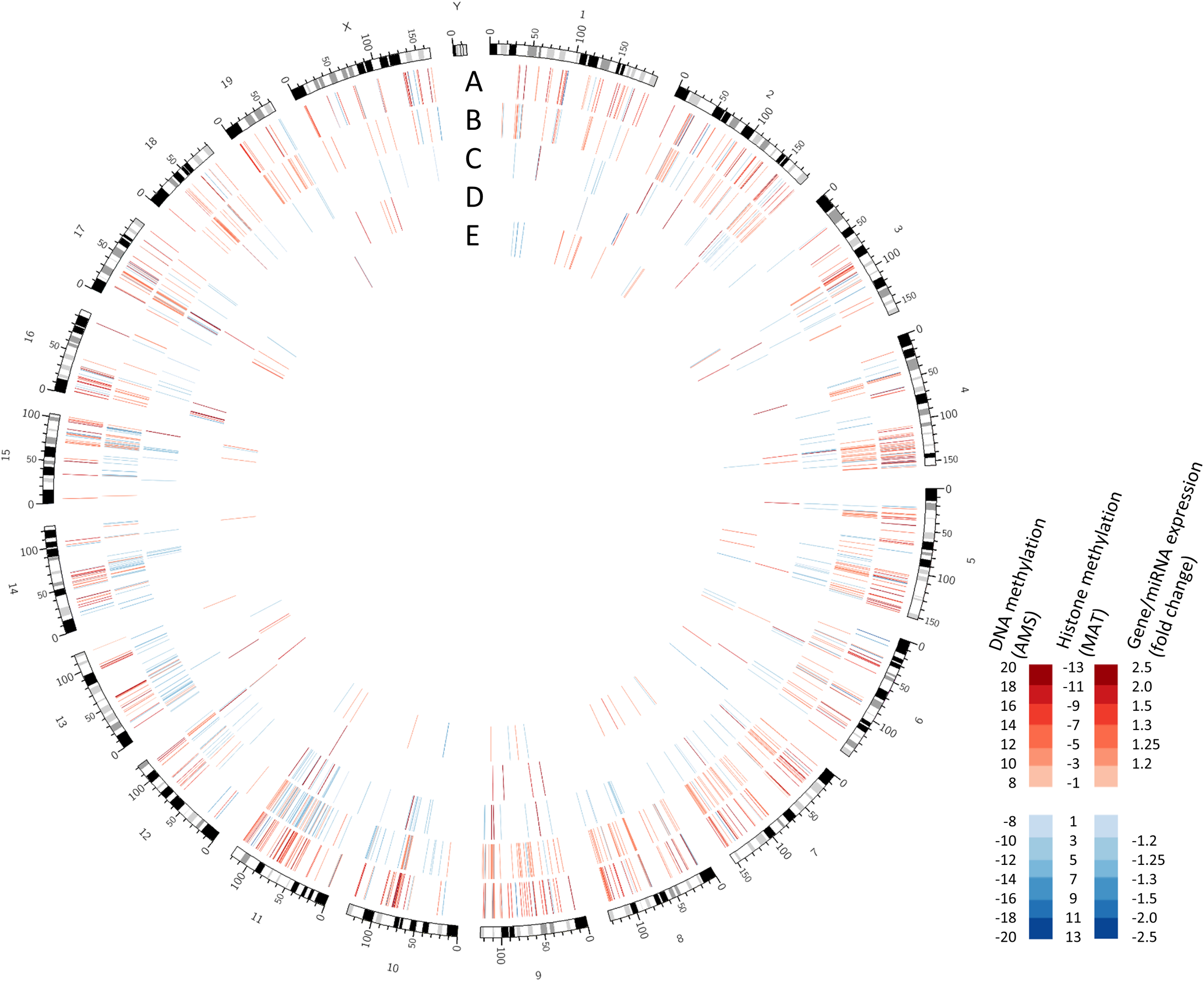
Global alterations to the adult hippocampus promoterome from Chater-Diehl et al. Tracks show alterations in: (A) DNA methylation (B) H3k27me3 (C) H3k4me3 (D) miRNA expression and (E) gene expression.
An Integrated Perspective
Lead author Eric Chater-Diehl shares, “When all three epigenetic methylations we looked at were grouped, we found enrichment of oxidative stress pathway genes. Redox genes were also our top gene expression group. Though few of the genes are shared between expression and epigenetic changes, the same cellular processes were implicated. These types of relationships are missed when examining individual marks or genes.”
Notably, oxidative stress is triggered immediately by prenatal alcohol exposure, which makes the profile observed mechanistically interesting, as alterations to oxidative stress systems are maintained into adulthood long after the early life exposure. Ultimately, this report highlights the potential of integrating diverse data sets as multiple lines of evidence that unearth alterations to different parts of the same complex systems.
Get a full view of this epigenetic landscape over at PLOS ONE, May 2016